Der Renner-Effekt
In Zusammenarbeit mit P. R. Bunker (Steacie Institute for Molecular Sciences, National Research Council of Canada, Ottawa), T. Hirano (Ochanomizu-Universität, Tokio, Japan), W. P. Kraemer (Max-Planck-Institut für Astrophysik, Garching) und anderen untersuchen wir, wie die Bahn- und Spindrehimpulse der Elektronen das Spektrum eines dreiatomigen Moleküls beeinflussen. Die entsprechenden Effekte nennt man allgemein den Renner-Effekt. Ursprünglich entwickelten wir das Computerprogramm RENNER zur Berechnung der Positionen und Intensitäten von Linien in einem Spektrum, dessen Übergänge innerhalb und zwischen den beiden Komponenten eines Renner-Zustandes stattfinden. Das RENNER-Programm ist in den letzten 15 Jahren für viele Moleküle, auch für freie Radikale und Molekülionen (insbesondere CH2+ und NH2+), erfolgreich eingesetzt worden. Die benötigten Potential- und Dipolmomentflächen werden mittels ab initio-Methoden berechnet. Die von RENNER vorhergesagten rovibronischen Spektren sind bei der Suche nach den entsprechenden experimentellen Spektren, und deren Interpretation, hilfreich gewesen.
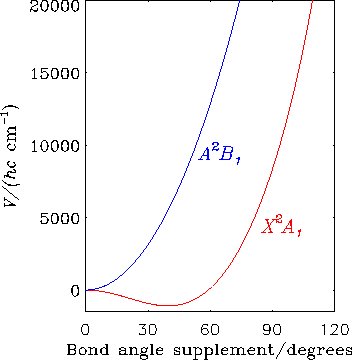
Die Abbildung zeigt wie die Elektronenzustände X2A1 und A2B1 des CH2+-Ions bei linearen Geometrien entartet werden; die Abszisse ist das Supplement des H-C-H Bindungswinkels. Diese beiden Elektronenzustände zeigen den Renner-Effekt.
Ende des zwanzigsten Jahrhunderts wurden Coulomb explosion imaging (CEI) Experimente für CH2+ durchgeführt. Basierend auf der Interpretation dieser Experimente wurde dann behauptet, dass die Wellenfunktionen der niedrigsten rovibronischen Zustände eine große nicht-adiabatische Komponente haben, viel größer als die Komponente, die vom Renner-Effekt herrührt. Wir berechneten anschließend die Energien der angeregten Elektronenzustände und fanden, in Übereinstimmung mit früheren Literaturergebnissen, dass die angeregten Elektronenzustände in CH2+ bei so hohen Energien liegen (> 6 eV), dass nicht-adiabatische Kopplungen vernachlässigt werden können. Damit wir mit den CEI-Ergebnissen vergleichen konnten, haben wir auch die Boltzmann-gemittelte Wahrscheinlichkeitsverteilung für den Knickwinkel berechnet. Wir benutzten für diese Berechnung unsere bereits ermittelten ab initio Potentialflächen für die Renner-wechselwirkenden X/A Zustände. In der Berechnung der rovibronischen Wellenfunktionen beschreiben wir die Renner-Wechselwirkung vollständig. Diese ab initio Berechnung erzeugt eine Wahrscheinlichkeitsverteilung die wesentlich "schmaler" als die CEI-gemessene Verteilung ist. Nach der gemessenen Verteilung kann das Molekül sich über ein erheblich breiteres Winkelintervall bewegen, als die Theorie vorhersagt. Es war lange unklar, warum Theorie und Experiment so weit aus einander lagen. Durch eine Re-Analyse der experimentellen CEI-Daten stellte es sich dann heraus, dass es bei der ursprünglichen Analyse eine Ungenauigkeit gegeben hat. Die neu interpretierten experimentellen Daten waren in guter Übereinstimmung mit unserer Vorhersage.
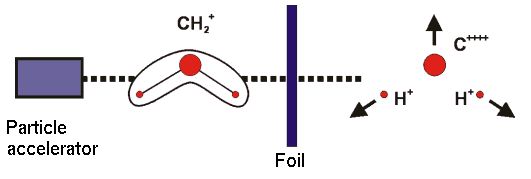
Die Abbildung zeigt das Prinzip eines CEI-Experiments. Molekülionen werden beschleunigt und durch eine Folie "geschossen". In der Folie verlieren sie viele Elektronen und das resultierende System ist sehr instabil. Es "explodiert" wegen abstoßender Coulombkräfte. Die Fragmente, die durch die Explosion entstehen, können detektiert und ihre kinetische Energien bestimmt werden. Mit der gewonnenen Information kann die molekulare Struktur im Augenblick der Explosion rekonstruiert werden.
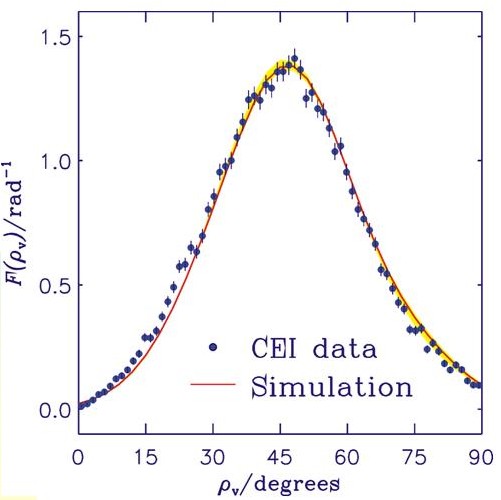
Die Abbildung zeigt die experimentellen CEI-Datenpunkte als gefüllte Kreise und das Ergebnis unserer Simulation als eine durchgezogene Kurve. Der Winkel ρv ist das Supplement des C-H-C Bindungswinkel. Die hier gezeigte Kurve beinhaltet die Effekte von sogenannten Folieneffekten (d.h. Verbreiterungseffekte, die auf die Wechselwirkung zwischen den Molekülionen und den Teilchen in der Folie zurückzuführen sind) auf die "molekül-interne" Kurve aus unserer Berechnung. Die Folieneffekte werden mittels Monte-Carlo-Methoden simuliert. Für die Einzelheiten des Experiments, siehe L. Lammich, Diploma thesis, Faculty of Physics and Astronomy, Ruprecht-Karls-Universität, Heidelberg, 2001, sowie L. Lammich, H. Buhr, H. Kreckel, S. Krohn, M. Lange, D. Schwalm, R. Wester, A. Wolf, D. Strasser, D. Zajfman, Z. Vager, L. Abril, S. Heredia-Avalos, and R. Garcia-Molina, Phys. Rev. A 69, 062904 (2004). Die Anregung, die CEI-Experimente zu re-analysieren, kam von unseren theoretischen Ergebnissen. Wir haben also hier einen der sehr seltenen Fällen, wo die Theorie das Experiment geändert hat.

Für das HO2 Molekül in den Elektronenzuständen X2A'' und A2A' haben wir weitere RENNER-Berechunungen durchgeführt. Die Abbildung zegt die Knickschwingungs-Potentialkurven für die beiden Elektronenzustände.
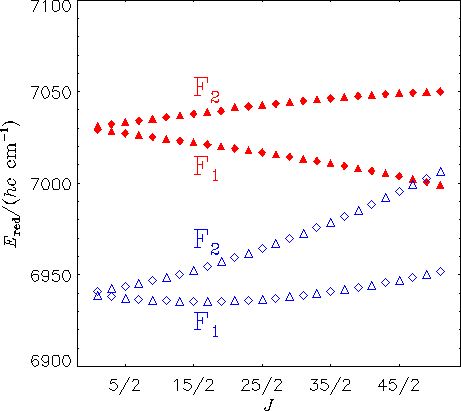
Das obige Diagramm zeigt reduzierte Energien für die Ka = 0 Zustände in den A(0,0,0) (gefüllte Dreiecke und Rauten) und X(1,1,2) (leere Dreiecke und Rauten) vibronischen Zuständen von HO2. Eine Raute stellt einen Zustand mit Plusparität [Symmetrie A' in Cs(M)] dar und ein Dreieck stellt einen Zustand mit Minusparität [Symmetrie A'' in Cs(M)] dar. Die theoretischen Ergebnisse zeigen, dass es eine lokale Störung der A(0,0,0) Niveaus in der Nähe von J = 51/2 gibt, in guter Übereinstimmung mit den experimentellen Beobachtungen von E. H. Fink und D. A. Ramsay [J. Mol. Spectrosc. 185, 304-324 (1997)]. Basierend auf unseren theoretischen Berechnungen kann der störende Zustand als X(1,1,2) identifiziert werden.
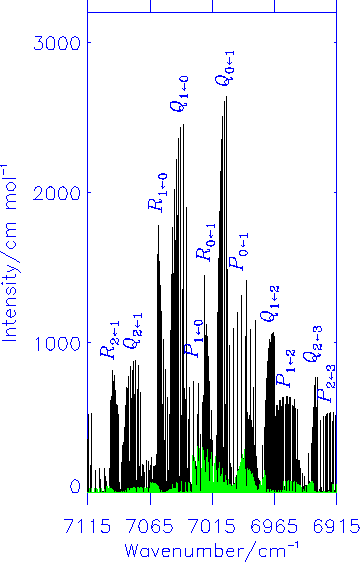
Zusätzlich fanden Fink und Ramsay [J. Mol. Spectrosc. 185, 304-324 (1997)] eine Anzahl von Linien mit anormaler Auswahlregel. Diese Übergänge verbinden anscheinend Zustände mit der gleichen Parität und dieses deutet an, dass sie magnetische Dipolübergänge sind. Unsere theoretische Berechnungen haben bestätigt, dass für den elektronischen Übergang X2A'' ← A2A' des HO2-Moleküls die magnetischen Dipolübergänge Intensitäten haben, die etwa 10% der Intensität der elektrischen Dipolübergänge errreichen. Das Diagramm zeigt das simulierte elektrische Dipol-Spektrum (schwarz) und das simulierte magnetische Dipol-Spektrum (grün).
Der doppelte Renner-Effekt
In den letzten Jahren haben wir uns mit dem "doppelten" Renner-Effekt in dreiatomigen Molekülen beschäftigt. Der doppelte Renner-Effekt entsteht, wenn, durch Isomerisierung, zwei lineare Geometrien, ABC und BCA, für das Molekül ABC zugänglich sind, und die elektronische Energie bei beiden linearen Geometrien zweifach Renner-entartet ist. Im Rahmen der Doktorarbeit von Tina Erica Odaka haben wir ein neues Computerprogramm, DR, entwickelt. Dieses Programm berechnet die rovibronischen Energien für ein dreiatomiges Molekül in einem Renner-entarteten Elektronenzustand. Vladlen V. Melnikov hat später das DR-Programm so erweitert, dass es jetzt auch rovibronische Absorptions- und Emissionsspektren simulieren kann.
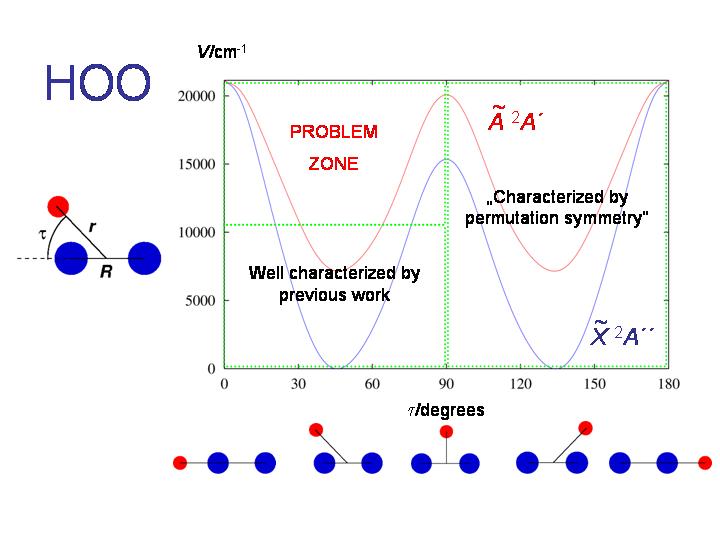
Wenn wir die RENNER-Beschreibung des HO2-Moleküls erweitern, liefert dieses Molekül ein Beispiel für den doppelten Renner-Effekt. Wir sehen in der Abbildung, dass HO2 bei Gleichgewicht gewinkelt ist. Es hat zwei äkvivalente Potentialminima, getrennt durch eine Barriere, die einer T-förmigen Molekülgeometrie entspricht. In der Nähe jeder der beiden Gleichgewichtsstrukturen finden wir eine lineare Struktur, bei der die beiden energetisch niedrigsten Elektronenzustände, X2A" und A2A', entartet werden.
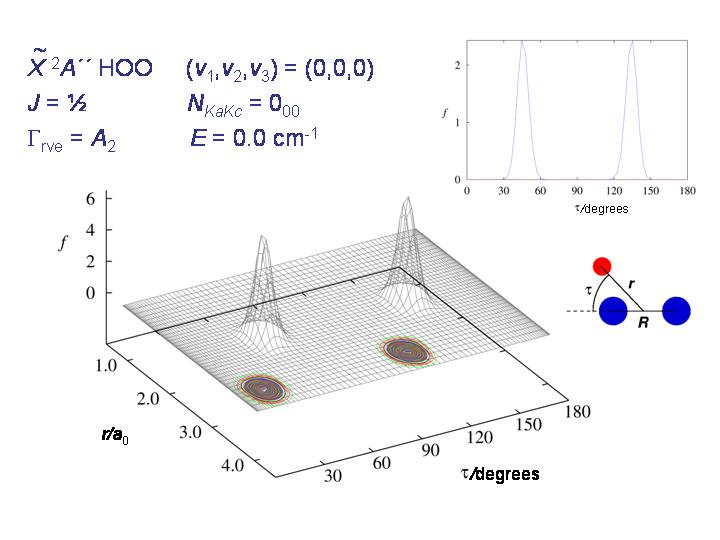
Die Abbildung zeigt für HO2 die Wahrscheinlichkeitsdichtefunktion für die Koordinaten r und τ (siehe Abbildung) im rovibronischen Grundzustand. Das Molekül ist in einer der beiden Potentialmulden "eingesperrt".
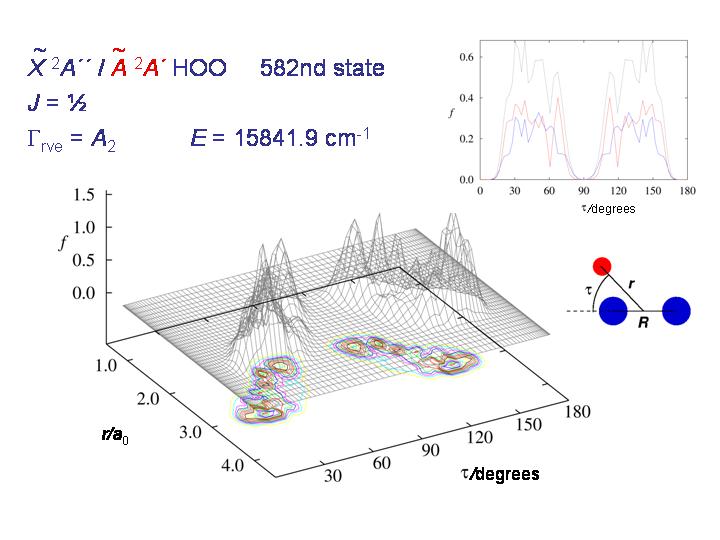
Bei höheren Energien kann das Molekül durch die Barriere "tunneln"; die Abbildung zeigt, wie die Wahrscheinlichkeitsverteilung sich "verbreitet".
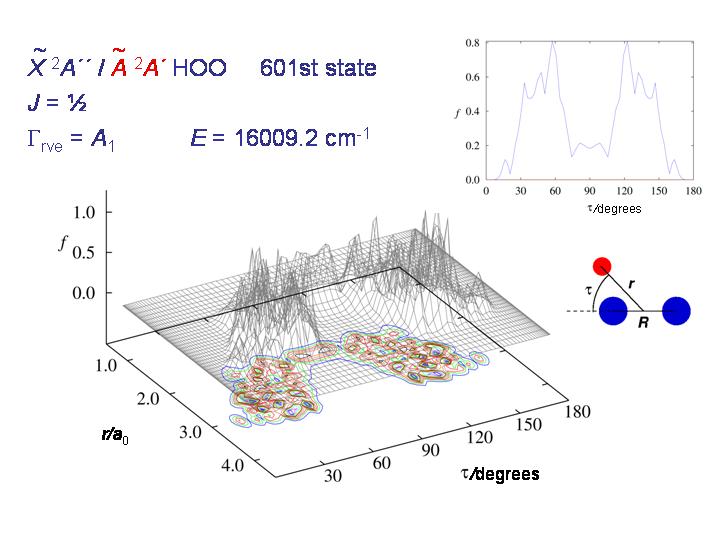
Bei noch höheren Energien kann das Moleküls sich über das ganze τ-Intervall von 0 bis 180 Grad bewegen und die Wahrscheinlichkeitsverteilung wird "chaotic and beautiful", wie T. E. Odaka es in ihrer Dissertation (2004) ausdrückte.
Mittels des DR-Programmes haben wir auch extensive theoretischen Untersuchungen der ersten angeregten Elektronenzustands, A2Π, des isomerisierenden doppel-Renner Moleküls MgNC/MgCN durchgeführt. Für dieses Molekül fanden wir Anzeichen für eine sogenannte Renner-getriebene Isomerisierung, wobei die Bewegung vom MgNC-Minimum zum MgCN-Minimum auf der A2Π-Potentialflächen von einem "electronischen Übergang" zwischen den beiden Komponenten des Π-Zustands begleitet wird.
Publikationen zum Renner-Effekt
(215) T. Hirano, U. Nagashima, and Per Jensen: Computational Molecular Spectroscopy of X2Π NCS: Electronic properties and ro-vibrationally averaged structure, J. Mol. Spectrosc. 346, 4–12 (2018). DOI: 10.1016/j.jms.2017.12.011
(214) J. Freund, S. C. Galleguillos Kempf, Per Jensen, U. Nagashima, and T. Hirano: Computational Spectroscopy of NCS in the Renner-degenerate electronic state X2Π, J. Mol. Spectrosc. 345, 31–38 (2018). DOI: 10.1016/j.jms.2017.11.010
(172) T. Hirano, U. Nagashima, G. Winnewisser, and Per Jensen: Electronic structures and rovibronically averaged geometries of the X6A' and Ã6A'' states of FeOH, J. Chem. Phys. 132, 094303/1-10 (2010).
(168) T. Hirano, P. R. Bunker, S. Patchkovskii, U. Nagashima, and Per Jensen: The Predicted Spectrum of FeOH in Its Renner-degenerate X6A' and Ã6A'' Electronic States, J. Mol. Spectrosc. 256, 45-52 (2009).
(166) V. V. Melnikov, Per Jensen, and T. Hirano: Calculation of Rovibronic Intensities for Triatomic Molecules in Double-Renner-degenerate Electronic States. Application to the X2A'' and Ã2A' Electronic States of HO2, J. Chem. Phys. 130, 224105/1-9 (2009).
(161) T. Hirano, R. Okuda, U. Nagashima, and Per Jensen: Computational Molecular Spectroscopy for X2Δ NiCN: Large Amplitude Bending Motion, J. Mol. Spectrosc. 250, 33-43 (2008).
(159) T. Hirano, R. Okuda, U. Nagashima, K. Tanaka, and Per Jensen: Ab Initio Molecular Orbital Study of Ground and Low-Lying Electronic States of NiCN, Chem. Phys., 346, 13-22 (2008).
(158) V. V. Melnikov, T. E. Odaka, Per Jensen, and T. Hirano: The Double Renner Effect in the X2A'' and Ã2A' Electronic States of HO2, J. Chem. Phys. 128, 114316/1-10 (2008).
(156) P. R. Bunker, W. P. Kraemer, S. N. Yurchenko, W. Thiel, C. F. Neese, J. L. Gottfried, and Per Jensen: New potential energy surfaces for the X and A states of CH2+, Mol. Phys. 105, 1369-1376 (2007).
(154) T. Hirano, M. Amano, Y. Mitsui, S. S. Itono, R. Okuda, U. Nagashima, and Per Jensen: A Theoretical Study of FeCN in the 6Δ Electronic Ground State, J. Mol. Spectrosc. 243, 267-279 (2007).
(153) T. Hirano, R. Okuda, U. Nagashima, Y. Nakashima, K. Tanaka, and Per Jensen: A theoretical study of BrCN+ in the 2Π electronic ground state: Large amplitude bending motion, J. Mol. Spectrosc. 243, 202-218 (2007).
(152) T. Hirano, R. Okuda, U. Nagashima, and Per Jensen: Ab Initio Molecular Orbital Study of Ground and Low-Lying Electronic States of CoCN, J. Chem. Phys. 127, 014303/1-7 (2007).
(151) T. Hirano, R. Okuda, U. Nagashima, and Per Jensen: A Theoretical Study of CoCN in the 3Φ Electronic Ground State, Mol. Phys. 105, 599-611 (2007).
(150) T. E. Odaka, V. V. Melnikov, Per Jensen, T. Hirano, B. Lang, and P. Langer: Theoretical Study of the Double Renner Effect for Ã2Π MgNC/MgCN: Higher Excited Rovibrational States, J. Chem. Phys. 126, 094301/1-9 (2007).
(146) T. E. Odaka, Per Jensen, and T. Hirano: The Double Renner Effect: A Theoretical Study of the MgNC/MgCN Isomerization in the Ã2Π Electronic State, J. Mol. Structure 795, 14-41 (2006).
(145) P. R. Bunker, R. Guérout, Z. J. Jakubek, Per Jensen, and S. N. Yurchenko: The rovibronic energies of the SiNSi radical in its X2Πg electronic state, J. Mol. Structure 795, 9-13 (2006).
(144) T. Hirano, R. Okuda, U. Nagashima, V. Špirko, and Per Jensen: A Theoretical Study of FeNC in the 6Δ Electronic Ground State, J. Mol. Spectrosc. 236, 234-247 (2006).
(128) S. Wu, Y. Chen, X. Yang, Y. Guo, Y. Liu, Y. Li, R. J. Buenker, and Per Jensen: Vibronic Transition Moments and Line Intensities in H2O+, J. Mol. Spectrosc. 225, 96-106 (2004).
(126) Per Jensen, W. P. Kraemer, and P. R. Bunker, Transition moments and NH2 cometary spectra, Mol. Phys. 101, 613-622 (2003).
(124) P. R. Bunker, W. P. Kraemer, Per Jensen, Y.-C. Lee, and Y.-P. Lee, The Matrix Isolation Spectrum of the CH2+ Ion, J. Mol. Spectrosc. 216, 419-423 (2002).
(123) T. E. Odaka, T. Hirano, and Per Jensen: A Theoretical Study of Ã2Π MgCN, J. Mol. Spectrosc. 216, 379-396 (2002).
(122) T. Hirano, K. Ishii, T. E. Odaka, and Per Jensen: A Theoretical Study of MgNC and MgCN in the X2Σ+ Electronic State, J. Mol. Spectrosc. 215, 42-57 (2002).
(120) T. E. Odaka, T. Hirano, and Per Jensen: An Ab Initio Study of the Ã2Π and the Ã2Π ← X2Σ+ Electronic Transition of MgNC, J. Mol. Spectrosc. 211, 147-161 (2002).
(119) Per Jensen, T. E. Odaka, W. P. Kraemer, T. Hirano, and P. R. Bunker: The Renner Effect in Triatomic Molecules with Application to CH2+, MgNC and NH2, Spectrochimica Acta Part A 58, 763-794 (2002).
(117) S. N. Yurchenko, Per Jensen, Y. Li, R. J. Buenker, and P. R. Bunker: The Near Ultraviolet Band System of Singlet Methylene, J. Mol. Spectrosc. 208, 136-143 (2001).
(116) P. R. Bunker, M.-C. Chan, W. P. Kraemer, and Per Jensen: Predicted Rovibronic Spectra of CH2+ and CD2+, Chem. Phys. Lett. 341, 358-362 (2001).
(115) Per Jensen, R. J. Buenker, J.-P, Gu, G. Osmann and P. R. Bunker: Refined Potential Energy Surfaces for the X2A'' and A2A' Electronic States of the HO2 Molecule, Can. J. Phys. 79, 641-652 (2001).
(111) G. Osmann, P. R. Bunker, W. P. Kraemer, and Per Jensen: Coulomb Explosion Imaging: The CH2+, H2O+ and NH2+ Ions as Benchmarks, Chem. Phys. Lett. 318, 597-606 (2000).
(108) G. Osmann, P. R. Bunker, W. P. Kraemer, and Per Jensen: Coulomb Explosion Imaging and the CH2+ Molecule, Chem. Phys. Lett., 309, 299-306 (1999).
(105) G. Osmann, P. R. Bunker, Per Jensen, R. J. Buenker, J.-P. Gu, and G. Hirsch: A Theoretical Investigation of the Renner Interactions and Magnetic Dipole Transitions in the A - X Electronic Band System of HO2, J. Mol. Spectrosc., 197, 262-274 (1999).
(103) J.-P. Gu, G. Hirsch, R. J. Buenker, M. Brumm, G. Osmann, P. R. Bunker and P. Jensen: A theoretical study of the absorption spectrum of singlet CH2, J. Mol. Struct., 517, 247-264 (2000).
(101) G. Osmann, P. R. Bunker, P. Jensen and W. P. Kraemer: An Ab Initio Study of the NH2+ Absorption Spectrum. J. Mol. Spectrosc. 186, 319 (1997)
(100) G. Osmann, P. R. Bunker, P. Jensen and W. P. Kraemer: A Theoretical Calculation of the Absorption Spectrum of CH2+. Chem. Phys. 225, 33 (1997).
(85) J.-P. Gu, R. J. Buenker, G. Hirsch, P. Jensen and P. R. Bunker: An ab initio calculation of the BH2- rovibronic energies: a very small singlet-triplet splitting. J. Mol. Spectrosc. 178, 172 (1996).
(79) M. Kolbuszewski, P. R. Bunker, W. P. Kraemer, G. Osmann and P. Jensen: An ab initio calculation of the rovibronic energies of the BH2 molecule. Mol. Phys. 88, 105 (1996).