Energie-Clusterbildung
Seit fast 20 Jahren ist es eine allgemein akzeptierte, experimentell nachgewiesene Tatsache, dass in den Schwingungszuständen des H2Se-Moleküls, bei hohen Werten der J- und Ka-Quantenzahlen, die Rotations-Schwingungsenergien Gruppen von jeweils vier fast-entarteten Niveaus, sogenannte Energieclusters, bilden. Realistische quantenmechanische Berechnungen haben gezeigt, dass die Moleküle H2S, H2Te und H2Po ähnliche Effekte aufweisen. In den 1990er Jahren beschäftigten wir uns mit der theoretischen Beschreibung der Energieclusters in dreiatomigen Molekülen. Diese theoretischen Arbeiten waren hauptsächlich Variationsrechnungen, das heißt, die Rotations-Schwingungsberechnungen wurden durch die Diagonalisierung einer Matrixdarstellung des Hamiltonoperators für Rotation und Schwingung erhalten. Die vier-fachen Clusters wurden ursprünglich durch klassische und semiklassische Theorie vorhergesagt und wir haben gezeigt, wie diese Vorhersagen vom Experiment und von quantenmechanischen Berechnungen bestätigt werden. Analyse der Rotations-Schwingungs-Wellenfunktionen liefert ein einfaches Bild der Rotationsbewegung in den Clusterzuständen: Das Molekül rotiert um eine seiner Bindungen, entweder im Uhrzeigersinn oder im Gegenuhrzeigersinn. Es gibt zwei Bindungen und zwei Möglichkeiten für die Rotationsrichtung. Dadurch entstehen vier äquivalente Situationen, einem vierfachen Cluster entsprechend.
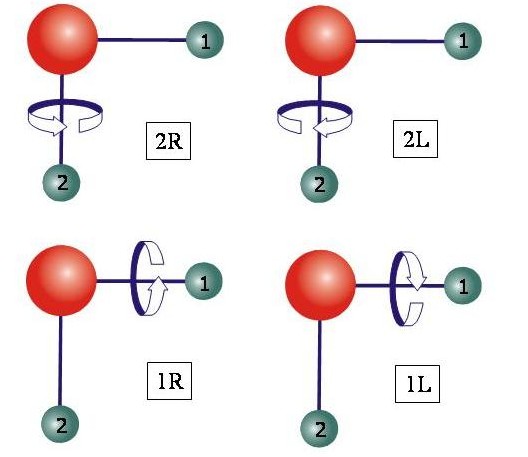
Das folgende Diagram illustriert die vier äquivalenten Situationen, die einen Clusterzustand eines dreiatomigen Moleküls darstellen.
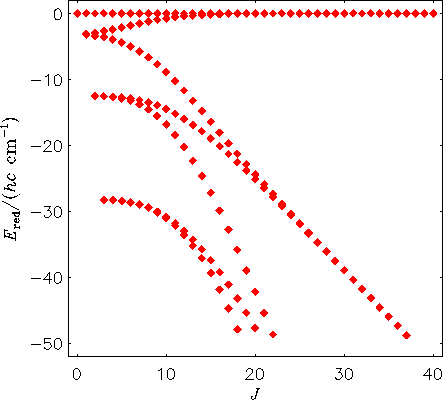
Wir plotten hier ein Energietermschema eines starren H2130Te-Moleküls. Bei diesem Molekül werden also Bindungslängen und Bindungswinkel konstant gehalten; die Werte entsprechen der Gleichgewichtsstruktur. Die Energiewerte sind relativ zur höchsten Energie bei jedem J-Wert geplottet.
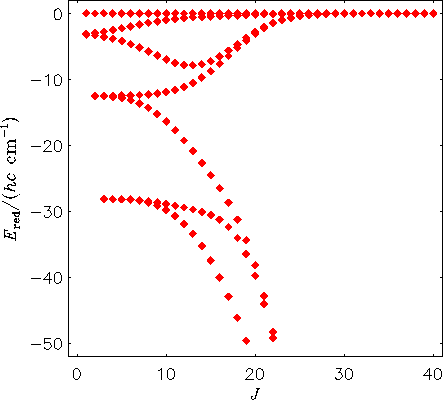
Das nächste Abbildung zeigt entsprechend das Termwertschema für das H2130Te-Molekül, berechnet mit dem MORBID-Programm direkt von der Potentialfunktion. Das heißt, die Schwingungsbewegung ist bei der Energieberechnung berücksichtigt.
Man sieht, dass die Zentrifugalverzerrungseffekte zur Bildung von
vierfach entarteten Energieclustern führen.
Am Anfang des 21. Jahrhunderts waren die Energieclusters in dreiatomigen Molekülen H2X experimentell nachgewiesen und in quantenmechanischen Variationsrechnungen theoretisch analysiert. Auch war bekannt, dass in sphärischen Kreiselmolekülen wie CH4 und CF4 Energie-Clusterbildung stattfindet. Es schien also wahrscheinlich, dass die "dazwischenliegenden" symmetrischen Kreiselmoleküle XH3 Energie-Clusterbildung aufweisen würden, aber diese Clusterbildung war weder experimentell noch theoretisch untersucht worden.
Als wir (in Zusammenarbeit mit Dr. Sergei N. Yurchenko, TU Dresden, und Prof. Walter Thiel, Max-Planck-Institut für Kohlenforschung, Mülheim/Ruhr) das Programm XY3 zur Berechnung der Rotations-Schwingungsenergien eines XH3-Moleküls in einem isolierten Elektronenzustand entwickelt hatten, haben wir dieses Programm eingesetzt, um eine mögliche Energie-Clusterbildung bei hoher Rotationsanregung im Phosphinmolekül PH3 theoretisch nachzuweisen.
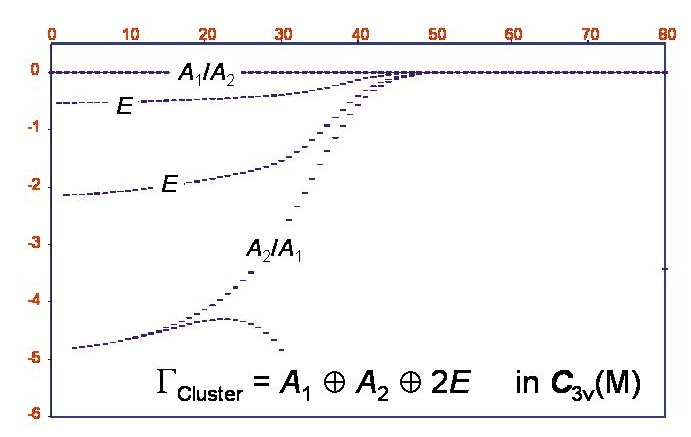
Rotations-Energietermverte des Phosphin-Moleküls im Schwingungsgrundzustand
Durch die XY3-Berechnungen konnten wir tatsächlich nachweisen, dass im Schwingungsgrundzustand des PH3-Moleküls bei hoher Rotationsanregung sechsfache Energieclustersgebildet werden. Die Abbildung zeigt ein Energietermschema für den Schwingungsgrundzustand von PH3; die Abszisse = J und die Ordinate ist der Termwert in cm-1 relativ zum höchsten Termwert für den gegebenen J-Wert. Der Clusterzustand ist insgesamt sechsfach entartet und entspricht der Symmetrie A1+A2+2E in der molekularen Symmetriegruppe C3v(M).
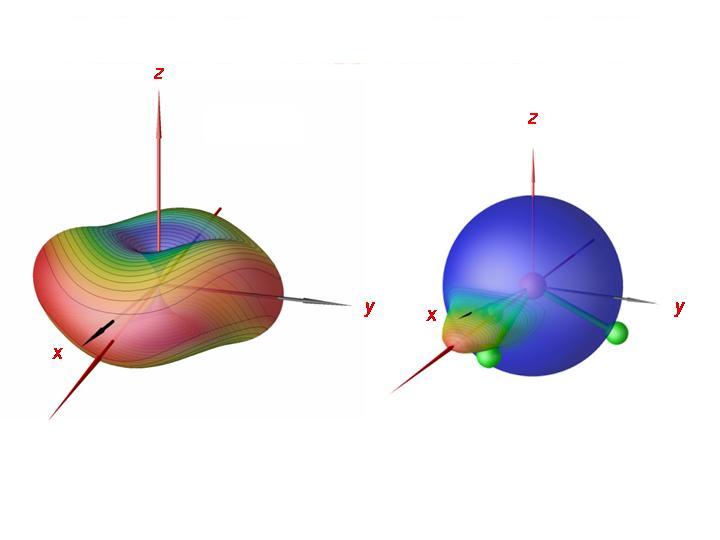
Durch Analyse der Rotations-Schwingungswellenfunktionen konnten wir ferner nachweisen, dass das PH3-Molekül um eine seiner Bindungen rotiert, entweder im Uhrzeigersinn oder im Gegenuhrzeigersinn. Es gibt jetzt drei Bindungen und zwei Möglichkeiten für die Rotationsrichtung. Dadurch entstehen sechs äquivalente Situationen, einem sechsfachen Cluster entsprechend. In der Abbildung haben wir rechts für einen Clusterzustand bei J = 80 die Wahrscheinlichkeitsdichte für die Richtung des Drehimpulsvektors auf eine Kugel relativ zum Molekül abgebildet. Man sieht, dass das Molekül annäherungsweise um einer seiner Bindungen rotiert. Links zeigen wir die semiklassische Rotationsenergiefläche bei J = 80. Die semiklassische Analyse bestätigt die quantenmechanischen Ergebnisse; das Molekül rotiert um eine seiner Bindungen.
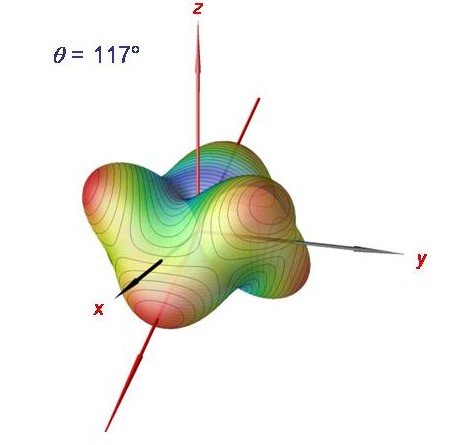
Rotationsenergiefläche des Phosphin-Moleküls bei J = 120.
Die semiklassische Analyse mittels Rotationsenergieflächen kann bis sehr hohe J-Werte durchgeführt werden. Hier zeigen wir die Phosphin-Rotationsenergiefläche für J = 120. Sie besitzt sechs äquivalente Maxima, entsprechend der sechs entarteten Clusterzustände.
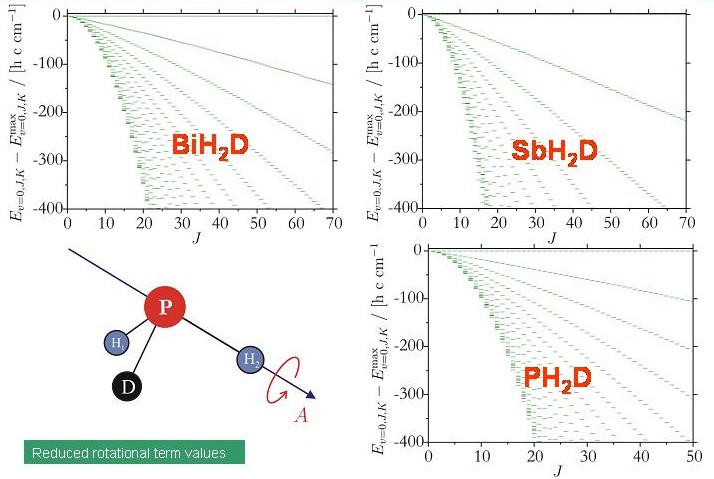
Mit der Entwicklung (in Zusammenarbeit mit Dr. Sergei N. Yurchenko, TU Dresden, und Prof. Walter Thiel, Max-Planck-Institut für Kohlenforschung, Mülheim/Ruhr) des Programmsystems TROVE (Theoretical ROtation-Vibration Energies) wurde es möglich, die Rotations-Schwingungsenergien eines willkürlichen Moleküls in einem isolierten Elektronenzustand durch eine Variationsrechnung zu bestimmen und wir konnten die Energie-Clusterbildung der deuterierten Moleküle XH2D und XHD2 (X = Bi, Sb, P) theoretisch untersuchen. Das Diagramm zeigt Energietermschemen für die untersuchten XH2D-Moleküle. Keine Energieclusters sind erkennbar.
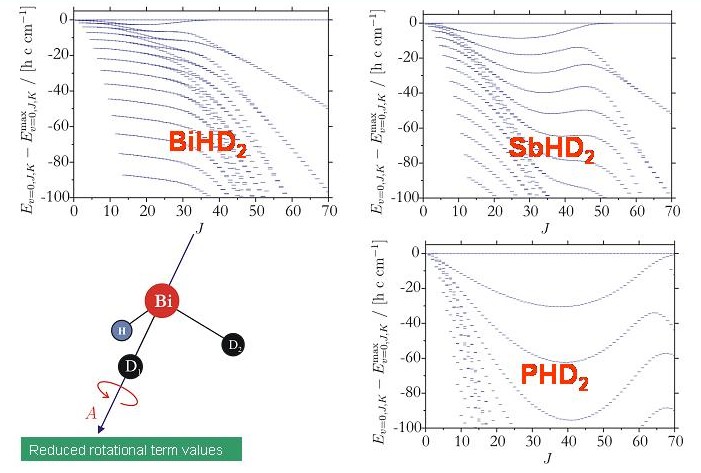
Hingegen bilden die Rotations-Schwingungsenergien der untersuchten XHD2-Moleküle vierfache Clusters, die in den Energietermschemen dieser Moleküle deutlich zu erkennen sind. Die Clusterbildung der XHD2-Moleküle ähnelt stark der Clusterbildung der H2X-Moleküle (siehe oben).
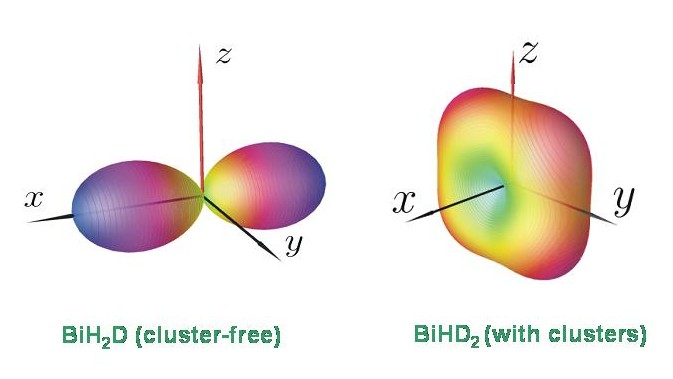
Es ist im Moment eine offene Frage, warum zum Beispiel BiHD2 Energieclusterbildung aufweist, aber BiH2D nicht. Die mit TROVE berechneten Energiestrukturen stimmen aber mit den Ergebnissen einer semiklassischen Rotations-Energiefläche-Analyse überein. Das Diagramm zeigt die Rotationsenergieflächen für BiH2D und BiHD2 bei J = 80. Die Rotationsenergiefläche für BiHD2 besitzt vier äquivalente Maxima, entsprechend einem vierfachen Cluster. Die Rotationsenergiefläche für BiH2D besitzt nur zwei äquivalente Maxima und dieses Molekül hat daher nur die zweifach entarteten Energieclusters, die für einen starren asymmetrischen Kreisel erwartet werden.
Veröffentlichungen zu Energieclustern
S. N. Yurchenko, R. I. Ovsyannikov, W. Thiel, and Per Jensen: Rotation-Vibration Energy Cluster Formation in XH2D and XHD2 Molecules (X = Bi, P, and Sb), J. Mol. Spectrosc. 256, 119-127 (2009).
S. N. Yurchenko, W. Thiel, and Per Jensen: Rotational energy cluster formation in XY3 molecules: Excited vibrational states of BiH3 and SbH3, J. Mol. Spectrosc. 240, 197-210 (2006).
S. N. Yurchenko, W. Thiel, Per Jensen, and P. R. Bunker: Rotation-vibration energy level clustering in the X2B1 ground electronic state of PH2, J. Mol. Spectrosc. 239, 160-173 (2006).
S. N. Yurchenko, W. Thiel, S. Patchkovskii, and Per Jensen: Theoretical Evidence for the Formation of Rotational Energy Level Clusters in the Vibrational Ground State of PH3, Phys. Chem. Chem. Phys. 7, 573-582 (2005).
P. R. Bunker and Per Jensen: Chirality in Rotational Energy Level Clusters, J. Mol. Spectrosc. 228, 640-644 (2004).
Per Jensen: An Introduction to the Theory of Local Mode Vibrations, Mol. Phys. 98, 1253-1285 (2000). Article prepared by invitation.
Per Jensen, G. Osmann, and I. N. Kozin: The Formation of Four-fold Rovibrational Energy Clusters in H2S, H2Se, and H2Te, in: "Advanced Series in Physical Chemistry", vol. 9, "Vibration-Rotational Spectroscopy and Molecular Dynamics" (D. Papousek, Ed., ISBN 981-02-1635-1), pp. 298-351, World Scientific Publishing Company, Singapore, 1997.
P. C. Gomez, L. F. Pacios, and Per Jensen: Fourfold Clusters of Rovibrational Energies in H2Po Studied with an ab initio Potential Energy Function, J. Mol. Spectrosc. 186, 99 (1997).
P. C. Gomez and Per Jensen: A Potential Energy Surface for the Electronic Ground State of H2Te Derived from Experiment, J. Mol. Spectrosc. 185, 282 (1997).