Simulation der rovibronischen Spektren kleiner Moleküle
In der Molekülspektroskopie wird Licht analysiert, das von Molekülen absorbiert oder ausgesandt wird. Dadurch werden Informationen über die Art der vorhandenen Moleküle und ihre Häufigkeit gewonnen, und zwar nicht nur im Labor, sondern auch in schwierig zugänglichen Umgebungen und weit entfernten Orten wie beispielsweise den oberen Schichten der Erdatmosphäre oder dem interstellaren Raum und den Atmosphären anderer Planeten und Kometen und kühler Sterne. Spektroskopische Methoden sind also ideal zur Fernerkundung ("remote sensing") und spielen eine wichtige Rolle in der Umweltforschung, so in der Erforschung der Erdatmosphäre sowie in der Astrophysik und –chemie.
Zur Unterstützung der Erforschung des interstellaren Raums, der äusseren Schichten von kühlen Sternen und der höheren Schichten der Erdatmosphäre durch Fernerkundungsexperimente (z.B. Radioastronomie) werden die rovibronischen Spektren kleiner Moleküle, die im Weltraum oder in der Erdatmosphäre vorhanden sind oder vorhanden sein könnten, simuliert. Diese Simulationen werden mit Methoden durchgeführt, die während der letzten 25 Jahre in der Arbeitsgruppe entwickelt oder mitentwickelt worden sind, insbesondere mit den Computerprogrammsystemen MORBID (Morse Oscillator Rigid Bender Internal Dynamics), RENNER, DR (Double Renner) und TROVE (Theoretical ROtation-Vibration Energies). Als Grundlage der Simulationen dienen ab initio Potential- und Dipolmomentflächen, die von Zusammenarbeitspartnern berechnet werden.

Als Beispiel zeigen wir hier das Rotationsspektrum im Schwingungsgrundzustand des HSOH-Moleküls, simuliert mit dem Programm TROVE bei einer Temperatur von 300 K.
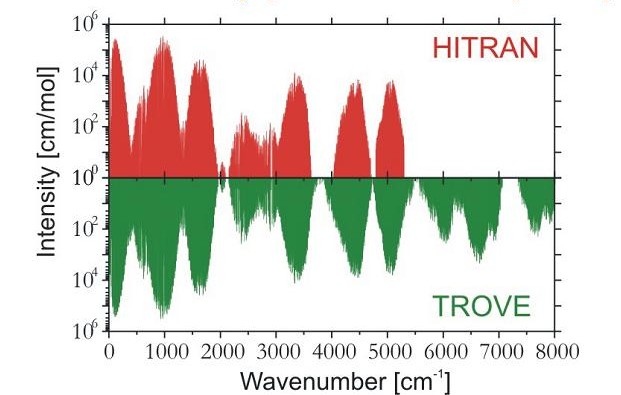
Mit der Entwicklung (in Zusammenarbeit mit Dr. Sergei N. Yurchenko, TU Dresden, und Prof. Walter Thiel, Max-Planck-Institut für Kohlenforschung, Mülheim/Ruhr) des Programmsystems TROVE (Theoretical ROtation-Vibration Energies) wurde es möglich, das Rotations-Schwingungsspektrum eines willkürlichen Moleküls in einem isolierten Elektronenzustand durch Variationsrechnungen zu simulieren. Das Programm wurde zunächst eingesetzt, um eine sogenannte Linienliste (das heißt, ein Katalog mit den Wellenzahlen und Intensitäten möglichst vieler Übergänge) für Ammoniak NH3 zu generieren. Der Zweck dieser sehr anspruchsvollen Berechnungen (die in Zusammenarbeit mit Dr. Yurchenko, Prof. Thiel und Prof. Jonathan Tennyson, University College London, UK, auf Rechern der University College London durchgeführt wurden) ist die Unterstützung von Beobachtungen und Analysen von Kometen und kühlen Sternen. Anfänglich wurde eine Ammoniak-Linienliste bei einer Temperatur von T = 300 K berechnet. Das Diagramm zeigt den Vergleich zwischen dieser Liste und den Einträgen der Spektroskopie-Datenbank HITRAN. Man erkennt, dass die theoretische Berechnung die beobachteten Linien in HITRAN sehr gut reproduziert und eine große Anzahl Linien, die experimentell nicht beobachtet worden sind, zusätzlich liefert.
Ende 2010 wurde eine Linienliste für NH3 bei T = 1500 K fertiggestellt; diese Liste kann bei der Analyse von Spektren kühler Sterne eingesetzt werden.
Veröffentlichungen zu Simulationen von Spektren
B. Ostojić, P. R. Bunker, P. Schwerdtfeger, B. Assadollahzadeh, and Per Jensen: The predicted spectrum of the hypermetallic molecule MgOMg, Phys. Chem. Chem. Phys. 13, 7546–7553 (2011).
A. Yachmenev, S. N. Yurchenko, Per Jensen, O. Baum, T. F. Giesen, and W. Thiel: Theoretical rotation-torsion spectra of HSOH, Phys. Chem. Chem. Phys. 12, 8387 - 8397 (2010).
S. N. Yurchenko, M. Carvajal, A. Yachmenev, W. Thiel, and Per Jensen: A theoretical-spectroscopy, ab-initio-based study of the electronic ground state of 121SbH3, J. Q. S. R. T. 111, 2279-2290 (2010).
A. Yachmenev, S. Yurchenko, I. Paidarová, Per Jensen, W. Thiel, and S. Sauer: Thermal averaging of the indirect nuclear spin-spin coupling constants of ammonia: the importance of the large amplitude inversion mode, J. Chem. Phys. 132, 114305/1-15 (2010).
T. Hirano, V. Derpmann, U. Nagashima, and Per Jensen: Large amplitude bending motion in CsOH, studied through ab initio-based three-dimensional potential energy functions, J. Mol. Spectrosc. 263, 150-159 (2010)
B. Ostojić, Per Jensen, P. Schwerdtfeger, B. Assadollahzadeh, and P. R. Bunker: The predicted infrared spectrum of the hyperberyllium molecule BeOBe in its X1Σg+and a3Σu+ electronic states, J. Mol. Spectrosc. 263, 21-26 (2010)
T. Hirano, U. Nagashima, G. Winnewisser, and Per Jensen: Electronic structures and rovibronically averaged geometries of the X6A' and Ã6A'' states of FeOH, J. Chem. Phys. 132, 094303/1-10 (2010).
S. N. Yurchenko, R. J. Barber, A. Yachmenev, W. Thiel, Per Jensen, and J. Tennyson: A variationally computed T=300 K line list for NH3, J. Phys. Chem. A 113, 11845-11855 (2009).
S. N. Yurchenko, A. Yachmenev, W. Thiel, O. Baum, T. F. Giesen, V. V. Melnikov, and Per Jensen: An ab initio calculation of the vibrational energies and transition moments of HSOH, J. Mol. Spectrosc. 257, 57-65 (2009).
R. I. Ovsyannikov, Per Jensen, M. Yu. Tretyakov, and S. N. Yurchenko: On the Use of the Finite Difference Method in a Calculation of Vibration-Rotation Energies, Optika i Spektroskopiya 107, 236-243 (2009) [in Russian]. English translation as Optics and Spectroscopy 107, 221-227 (2009).
T. Hirano, P. R. Bunker, S. Patchkovskii, U. Nagashima, and Per Jensen: The Predicted Spectrum of FeOH in Its Renner-degenerate X6A' and Ã6A'' Electronic States, J. Mol. Spectrosc. 256, 45-52 (2009). DOI: 10.1016/j.jms.2009.01.013
V. V. Melnikov, Per Jensen, and T. Hirano: Calculation of Rovibronic Intensities for Triatomic Molecules in Double-Renner-degenerate Electronic States. Application to the X2A'' and Ã2A' Electronic States of HO2, J. Chem. Phys. 130, 224105/1-9 (2009).
R. I. Ovsyannikov, W. Thiel, S. N. Yurchenko, M. Carvajal, and Per Jensen: PH3 revisited: Theoretical transition moments for the vibrational transitions below 7000 cm-1, J. Mol. Spectrosc. 252, 121-128 (2008).
R. I. Ovsyannikov, V. V. Melnikov, W. Thiel, Per Jensen, O. Baum. T. F. Giesen, and S. N. Yurchenko: Theoretical rotation-torsion energies of HSOH, J. Chem. Phys. 129, 154314/1-9 (2008).
R. I. Ovsyannikov, W. Thiel, S. N. Yurchenko, M. Carvajal, and Per Jensen: Vibrational energies of PH3 calculated variationally at the complete basis set limit, J. Chem. Phys. 129, 044309/1-8 (2008).
S. N. Yurchenko, W. Thiel, M. Carvajal, and Per Jensen: Ab initio potential energy surface, electric dipole moment, polarizability tensor, and theoretical rovibrational spectra in the electronic ground state of 14NH3+, Chem. Phys., 346, 146-159 (2008).
S. N. Yurchenko, W. Thiel, and Per Jensen: Theoretical ROVibrational Energies (TROVE): A robust numerical approach to the calculation of ro-vibrational energies for polyatomic molecules, J. Mol. Spectrosc. 245, 126-140 (2007).
P. R. Bunker, W. P. Kraemer, S. N. Yurchenko, W. Thiel, C. F. Neese, J. L. Gottfried, and Per Jensen: New potential energy surfaces for the X and A states of CH2+, Mol. Phys. 105, 1369-1376 (2007).
S. N. Yurchenko, M. Carvajal, W. Thiel, and Per Jensen: Ab initio dipole moment and theoretical rovibrational intensities in the electronic ground state of PH3, J. Mol. Spectrosc. 239, 71-87 (2006).
P. R. Bunker, R. Guérout, Z. J. Jakubek, Per Jensen, and S. N. Yurchenko: The rovibronic energies of the SiNSi radical in its X2Πg electronic state, J. Mol. Structure 795, 9-13 (2006). DOI: 10.1016/j.molstruc.2006.02.014
Z. J. Jakubek, P. R. Bunker, M. Zachwieja, S. G. Nakhate, B. Simard, S. N. Yurchenko, W. Thiel, and Per Jensen: A Dispersed Fluorescence and Ab Initio Investigation of the X2B1 and Ã2A1 Electronic States of the PH2 Molecule, J. Chem. Phys. 124, 094306/1-5 (2006).