Numerical Methods and Simulations
In the natural sciences, attempts are made to describe natural phenomena using mathematical methods, that is, through mathematical equations. One of the major challenges is the realisation that, whilst a great many natural processes can in principle be described in the language of mathematics, the resulting equations cannot be solved analytically – that is, with mathematical precision. If, in principle, no exact solution to a specific problem is possible, one must resort to approximations and simplifications. We attempt to approximate the exact solution using clever approximation techniques. As these discrete approximation techniques are based on specific calculations involving specific numbers, they are called numerical methods.
In theoretical chemistry, numerical methods have long been the primary tool, but they are also becoming increasingly important in physical chemistry.
Classic problems in this field include chemical kinetics, for example the analysis and simulation of the time-dependent behaviour of complex chemical reactions, and the coupling of such kinetic calculations with atmospheric transport models.
More recently, our research group has been developing new numerical methods, primarily for method development in atmospheric pressure ionisation (API) mass spectrometry. Typical research questions in this area include the characterisation of gas flows (fluid dynamics), the motion of ions at atmospheric pressure, and the description of typical reaction mechanisms in AP ion sources.
Simulation of gas flow and ion motion at atmospheric pressure

Simulation of gas flows in an AP ion source (simulation conducted in collaboration with IST-Aachen)
The motion of ions at atmospheric pressure is not – as in a vacuum – determined solely by the electric fields present, but also depends heavily on the prevailing flow dynamics due to the intense interaction with the surrounding gas.
In order to describe the motion of ions in atmospheric-pressure ion sources and similar systems, models of fluid dynamics (Computational Fluid Dynamics – CFD) are therefore also required. We develop simpler flow models ourselves within our research group, whilst for more complex CFD calculations we collaborate with partners such as the Institute for Jet Propulsion and Turbo Machinery at RWTH Aachen University.
Based on these simulations, calculations of discrete ion trajectories (particle tracing) or ion distribution (using finite element methods (FEM) or finite volume methods) can then be carried out.

Simulation von Ionentrajektorien unter Atmosphärendruck
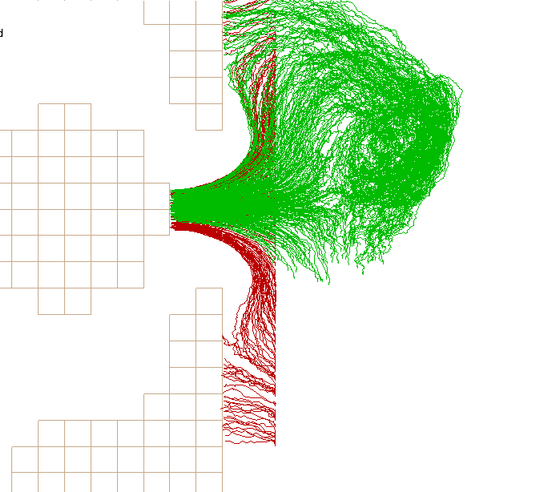
However, as such numerical simulations do not necessarily yield solutions that are valid in a physical sense, verifying the results is just as important a task as the simulation itself.
One way of verifying flow simulations is Particle Image Velocimetry (PIV), a method we have already successfully employed through our partnership with RWTH Aachen University.
Simulation of reaction kinetics
Kinetic modelling is used in physical and theoretical chemistry, amongst other things, to elucidate the reaction mechanisms of ion-molecule reactions, such as those occurring in atmospheric-pressure ion sources. The time-dependent behaviour of the reactions involved in ion transformation processes is typically described as a system of differential equations. To solve such a system of equations, one need only know the reaction rate constants of the reactions involved and the concentrations of the reactants. The concentrations of the reaction products are then calculated for a specific reaction time. These are subsequently compared with the signal intensities obtained from the mass spectrum in order to confirm or rule out the proposed reaction mechanism.
Kinetic Monte Carlo simulations / Simulation of reacting ion flows
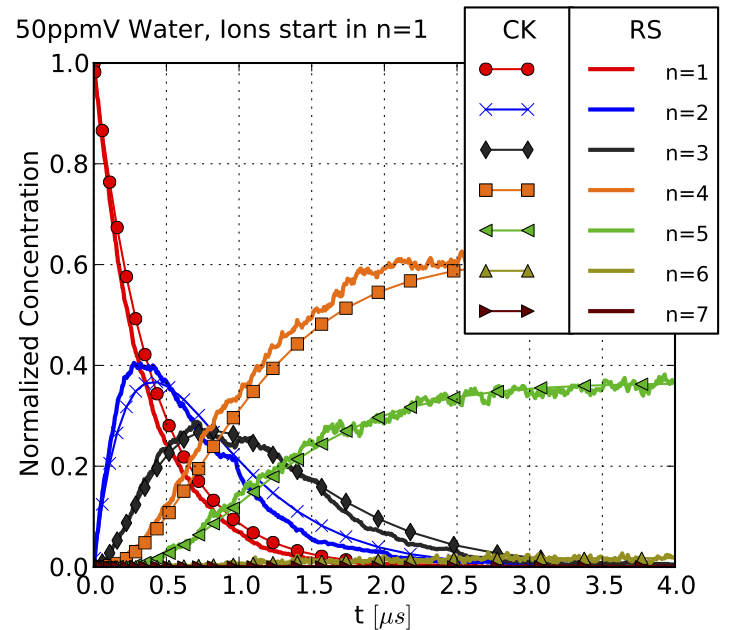
Simulation of the Temporal Evolution of Proton-Bound Water Clusters in AP Ion Mobility Spectroscopy (IMS)
The numerical description of ion transport in an electric field and under the influence of a flow of the surrounding gas can be combined with the simulation of the chemical reaction dynamics of the ions.
To this end, we have extended existing numerical models (SIMION / SDS) to include a corresponding module. Describing reactive ions in the gas phase within a computer model requires knowledge of the relevant chemical reactions that an ion may undergo. Consequently, a complete simulation of ion dynamics under atmospheric pressure conditions requires detailed knowledge, which must be obtained through prior research.
We have set up our own website for the development and exchange of information relating to the simulation code (Reaction Simulation – RS):
Ab initio method

Transient state of proton transfer from toluene to trimethylamine
In our research group, ab initio methods are used to verify reaction mechanisms. The free enthalpy of a reaction indicates whether the reaction is thermodynamically feasible. The activation energy indicates whether a reaction is kinetically constrained. Ab initio methods can be used to calculate molecular properties. In addition to properties such as vibrational frequencies and bond lengths, these also include the formation enthalpies of the molecules. The free reaction enthalpies can be calculated from the formation enthalpies, thereby confirming or refuting possible reactions. It is also possible to determine the geometry and energy of a reaction’s transition state, thereby obtaining information not only about the activation energy but also about the course of the reaction. The graphic/animation shows the transition state of proton transfer from toluene to trimethylamine.